

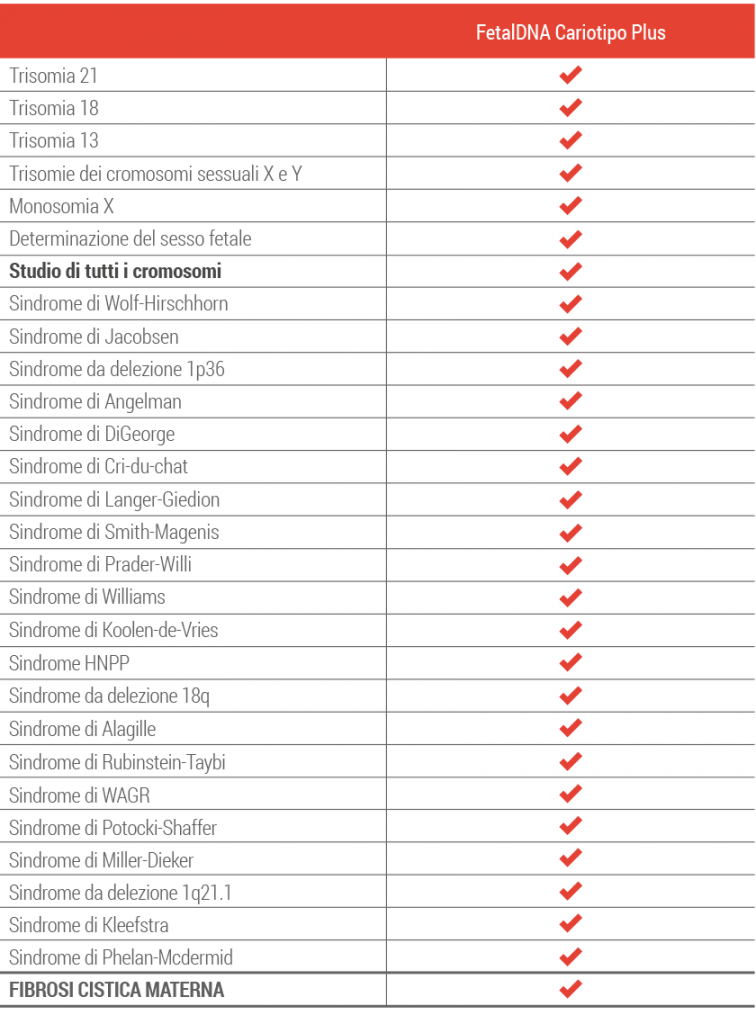
FetalDNA Cariotipo Plus è un test non invasivo di screening prenatale (NIPT) che prevede l’analisi del DNA fetale libero circolante su sangue materno, per rilevare informazioni riguardo a tutto il corredo cromosomico del feto e ad alterazioni strutturali associate a diverse condizioni patologiche con risultati paragonabili al cariotipo fetale ottenuto mediante tecniche diagnostiche invasive (amniocentesi e villocentesi).
Su richiesta è possibile ottenere gratuitamente il fattore RH del nascituro.
Ogni individuo presenta 2 copie di ciascun cromosoma. Le aneuploidie sono anomalie numeriche dei cromosomi. In particolare con trisomia si intende la presenza di un cromosoma in più a carico di una determinata coppia cromosomica, mentre con la monosomia si riscontra un solo cromosoma anziché 2.
Le aneuploidie studiate dal FetalDNA sono le più importanti e tra le più comuni che possono interessare il feto.
Trisomia del cromosoma 21
È l’aneuploidia più comune e si riferisce alla presenza di una copia in più del cromosoma 21. Questa sindrome è conosciuta come sindrome di Down e rappresenta, con un’incidenza di circa 1/650 nati, la forma più comune di ritardo mentale.
Trisomia del cromosoma 18
È la seconda aneuploidia più comune e si riferisce alla presenza di una copia in più del cromosoma 18. Questa sindrome è conosciuta come sindrome di Edwards ed è associata ad un elevato rischio di abortività. La sua incidenza si stima sia presente in circa 1/5000 nati.
Trisomia del cromosoma 13
È causata da una copia in più del cromosoma 13 ed è nota anche come sindrome di Patau. È associata ad una elevata abortività; i neonati presentano diverse condizioni patologiche che sono spesso causa di decessi durante l’infanzia. Si stima abbia un’incidenza di circa 1/16000nati.
Sindrome di DiGeorge (delezione 22q11.2)
È dovuta alla perdita (delezione) di una porzione del cromosoma 22. I bambini colpiti da sindrome di DiGeorge possono mostrare sviluppo incompleto del timo e delle ghiandole paratiroidi, cardiopatie congenite e anomalie del viso. Ha una incidenza di circa 1/3500 nati vivi.
Sindrome Cri-du-chat (delezione 5p)
È dovuta dalla delezione di una regione più o meno estesa del braccio corto del cromosoma 5. È caratterizzata da ritardo psicomotorio, microcefalia, anomalie del volto e dall’emissione di un pianto molto tipico (simile al miagolio di un gatto) durante la prima infanzia. Si stima abbia un’incidenza che varia da circa 1/15000 a 1/50000 nati vivi.
Sindrome di Prader-Willi
È una patologia molto eterogenea determinata da delezioni sul cromosoma 15. È caratterizzata da anomalie ipotalamico-pituitarie associate a grave ipotonia nel periodo neonatale e nei primi due anni di vita oltre all’ insorgenza di iperfagia che può evolvere nel rischio di obesità patologica durante l’infanzia e nell’età adulta, è spesso causa di difficoltà di apprendimento e disturbi comportamentali associati a problemi psichiatrici gravi e ritardo nello sviluppo psicomotorio. Ha una incidenza di circa 1/25.000 nati vivi.
Aneuploidie dei cromosomi sessuali
Sono anomalie che interessano i cromosomi sessuali XY e che possono causare nei neonati difficoltà di linguaggio, motori e/o di apprendimento. La più comune di questa classe di aneuploidie è la sindrome di Turner o monosomia legata al cromosoma X che colpisce le donne in cui è presente una sola copia del cromosoma X ed ha una incidenza di circa 1/2700 nati.
Altre aneuplodie riscontrabili con il FetalDNA Cariotipo Plus sono la Sindrome Klinefelter e la Sindrome di Jacobs.
Sindrome di Angelman
La sindrome di Angelman (AS) è una malattia neurologica, di origine genetica, caratterizzata da grave ritardo mentale e dismorfismi facciali caratteristici. La sua prevalenza è stimata tra 1/10.000 e 1/20.000. I pazienti appaiono normali alla nascita. Nei primi 6 mesi di vita possono manifestarsi disturbi dell’alimentazione e ipotonia, seguiti da ritardo dello sviluppo tra i 6 mesi e i 2 anni. In genere i sintomi caratteristici della AS si manifestano a partire dal primo anno di vita, con grave ritardo mentale, assenza del linguaggio, crisi di riso associate a movimenti stereotipati delle mani, microcefalia, macrostomia, ipoplasia mascellare, prognatismo e disturbi neurologici con andatura da ‘burattino’, atassia e attacchi epilettici associati ad anomalie specifiche all’elettroencefalogramma (EEG; attività delta trifasica con picchi nelle regioni frontali).
Sindrome di Wolf-Hirshhorn
La sindrome di Wolf-Hirshhorn (WHS) è una malattia dello sviluppo caratterizzata da segni craniofacciali caratteristici, ritardo della crescita prenatale e postnatale, deficit cognitivo, grave ritardo dello sviluppo psicomotorio, convulsioni e ipotonia.
Si osservano marcato ritardo della crescita prenatale, lentezza costante nel guadagno del peso postnatale, una facies tipica ad “elmo da guerriero greco” (radice del naso larga che continua sulla fronte) molto più evidente prima della pubertà, microcefalia, fronte alta con glabella prominente, ipertelorismo, epicanto, sopracciglia molto arcuate, filtro corto, bocca rivolta verso il basso, micrognatia, orecchie poco formate con fossette/appendici e, in alcuni casi, labiopalatoschisi. Sono presenti anomalie scheletriche, come la cifosi o scoliosi con malformazione dei corpi vertebrali, costole fuse o accessorie, piedi torti e schisi delle mani. È presente ipotonia con ipoplasia delle masse muscolari, che può associarsi a difficoltà alimentari e contribuire al ritardo della crescita. Il ritardo dello sviluppo è grave: molti pazienti non imparano a controllare lo sfintere, a mangiare e vestirsi autonomamente, e meno del 50% cammina, con o senza sostegni, tra i 2 e i 12 anni di età. Il deficit cognitivo è moderato-grave, di rado lieve. Il linguaggio si limita a suoni gutturali o bisillabici e, in alcuni casi, a semplici frasi. In oltre il 95% dei pazienti si osservano convulsioni a esordio tra il periodo neonatale e i 36 mesi, spesso scatenate dalla febbre.
Sindrome di Jacobsen
La sindrome di Jacobsen è una malattia da geni contigui con anomalie congenite multiple/ritardo mentale (MCA/MR), causata dalla delezione parziale del braccio lungo del cromosoma 11. Sono stati pubblicati più di 200 casi. La prevalenza è stimata in circa 1/100.000 nati, con un rapporto femmine/maschi di 2:1. I segni clinici più comuni sono il ritardo di crescita pre- e post-natale, il ritardo psicomotorio e i dismorfismi facciali che sono caratteristici (deformità del cranio, ipertelorismo, ptosi, coloboma, rime palpebrali rivolte verso il basso, epicanto, sella nasale ampia, naso corto, labbra a forma di V, orecchie piccole, a bassa attaccatura e retroruotate). Alla nascita sono di solito presenti anomalie funzionali delle piastrine, trombocitopenia o pancitopenia. I pazienti hanno spesso cardiopatie, difetti dei reni, del tratto gastrointestinale, dei genitali, del sistema nervoso centrale e dello scheletro. Possono essere presenti anche anomalie degli occhi, dell’apparato uditivo, immunologiche e ormonali.
Sindrome di Langer-Giedion
La sindrome di Langer-Giedion o sindrome tricorinofalangeale tipo 2 è caratterizzata da deficit cognitivo, associato a varie anomalie, compresa la cute ridondante, le esostosi cartilaginee multiple, la facies caratteristica e le epifisi falangeali ‘a cono”. La gravità e il numero di queste anomalie variano nei diversi pazienti. I dismorfismi facciali comprendono il naso globoso, il filtro ampio e prominente, il labbro superiore sottile, le orecchie ‘a cavolfiore”, i capelli radi e l’ipoplasia mandibolare. Sono stati descritti anche ritardo di crescita, microcefalia, ipotonia e problemi uditivi. Le esostosi si localizzano prevalentemente sulle estremità delle ossa lunghe e possono causare dolore, rigidità funzionale o deformazione. Le esostosi e le epifisi ‘a cono” compaiono nei primi 5 anni di vita, quando sono anche frequenti le infezioni delle vie respiratorie. La prevalenza non è nota. Si trasmette come carattere autosomico dominante, ma sono stati descritti soprattutto casi sporadici.
Sindrome da delezione 1p36
La sindrome da delezione 1p36 è un’anomalia cromosomica con dismorfismi facciali caratteristici, ipotonia, ritardo dello sviluppo, deficit cognitivo, convulsioni, cardiopatie, sordità e ritardo della crescita a esordio prenatale.
I pazienti condividono dismorfismi craniofacciali caratteristici con sopracciglia diritte, occhi infossati, ponte/sella nasale larga e piatta, ipoplasia mediofacciale, filtro lungo, mento appuntito e, spesso, fontanella anteriore grande con chiusura tardiva (>3 cm alla nascita), microbrachicefalia, orecchie anomale, ruotate posteriormente, a basso impianto. Sono anche caratteristici la brachidattilia, la campodattilia e i piedi corti. Quasi tutti i pazienti presentano ipotonia congenita, che concorre alle difficoltà all’alimentazione, al ritardo dello sviluppo motorio e della motilità fine, al ritardo o all’assenza del linguaggio. In tutti i pazienti è presente ritardo mentale variabile.
È considerata una delle più comuni sindromi da delezione cromosomica, con un’incidenza di 1/5.000-10.000 nati vivi. Colpisce in uguale misura entrambi i sessi, in tutte le etnie.
Sindrome di Williams
La sindrome di Williams è una malattia genetica rara caratterizzata da disturbi dello sviluppo, associati, nel 75% dei casi, a cardiopatie (di solito stenosi sopravalvolare dell’aorta, SSA), ritardo psicomotorio, dismorfismi facciali caratteristici ed un profilo cognitivo e comportamentale specifico. Questi bambini presentano una facies caratteristica con setto nasale appiattito e punta del naso globosa, bocca larga, con labbro inferiore anteverso, guance prominenti, edema periorbitale, epicanto e, spesso, iride ‘a stella’. Con l’età, il viso diventa più stretto e assume lineamenti grossolani. Il profilo cognitivo è caratterizzato da deficit visuo-spaziale, che contrasta con le buone capacità di linguaggio. I bambini affetti presentano un comportamento ipersociale e interagiscono bene con le altre persone. Sono molto sensibili al rumore e hanno buone capacità musicali.
Sindrome di Koolen-de Vries
La monosomia 17q21.31 (sindrome da microdelezione 17q21.31) è un’anomalia cromosomica caratterizzata da ritardo dello sviluppo, ipotonia nell’infanzia, dismorfismi facciali e comportamento amichevole/amabile. La prevalenza della sindrome è stimata in circa 1/16.000. I dismorfismi facciali sono caratterizzati da fronte alta e ampia, facies allungata, rime palpebrali rivolte verso l’alto, epicanto, forma anomala del naso (tubulare o a forma di pera), punta del naso globosa, orecchie grandi e prominenti e labbro inferiore anteverso. Sono frequenti le anomalie della pigmentazione e della consistenza dei capelli.
Neuropatia ereditaria con suscettibilità alle paralisi da pressione (HNPP)
La neuropatia ereditaria con suscettibilità alle paralisi da pressione (HNPP) è una malattia ereditaria dei nervi periferici con mononeuropatia ricorrente, di solito scatenata da blande attività fisiche. In rari casi, la HNPP può esordire con una plessopatia brachiale con paralisi e perdita sensoriale dolorosa monolaterale del braccio. Di rado si osservano lievi anomalie della funzione dei nervi cranici. In alcuni casi, mancano i riflessi tendinei profondi e sono presenti piedi cavi. Il fenotipo della HNPP spesso evolve in una polineuropatia motoria sensitiva simmetrica nei pazienti anziani.
Sindrome da delezione 18q
Le persone con la delezione prossimale del braccio lungo del cromosoma 18, hanno un cromosoma 18 intatto, ma l’altro è mancante di una parte di dimensioni variabili che può influenzare il loro apprendimento e sviluppo fisico. La maggior parte delle difficoltà cliniche sono probabilmente causate dalla presenza di una sola copia ( anziché due) di un certo numero di geni. Comunque, gli altri geni del bambino e la sua personalità lo aiutano a determinare il suo futuro sviluppo, i bisogni e le capacità.
Sindrome di Alagille
La sindrome di Alagille (AGS) è caratterizzata da colestasi cronica da paucità dei dotti biliari interlobulari, stenosi periferica dei rami dell’arteria polmonare, anomalie dei segmenti vertebrali, facies caratteristica, embriotoxon posteriore/anomalie del segmento anteriore, retinite pigmentosa e displasia renale. La prevalenza stimata è di circa 1/70.000. Nei neonati la malattia può manifestarsi associata a ittero prolungato con iperbilirubinemia e/o segni e sintomi cardiaci. Le anomalie cardiache includono atresia o stenosi polmonare, difetti del setto atriale e/o ventricolare, tetralogia di Fallot, dotto arterioso pervio. La colestasi è associata a iperbilirubinemia coniugata, epatosplenomegalia, ipercolesterolemia, ipertrigliceridemia e coagulopatia. Possono verificarsi anche prurito e xantomi. Tra le anomalie scheletriche minori si osservano emivertebre a farfalla (in circa il 50% dei casi), radio, ulna e falangi corti. La facies caratteristica, se presente, è evidente sin dalla nascita e è caratterizzata da fronte prominente, occhi infossati, rime palpebrali oblique rivolte verso l’alto, ipertelorismo, sella nasale appiattita e mento appuntito. Le anomalie oftalmiche includono embriotoxon posteriore (75% dei casi), anomalia di Axenfeld (si veda questo termine), retinite pigmentosa, anomalie pupillari e del disco ottico. In alcuni casi si osservano ritardo della crescita e dello sviluppo e malassorbimento dei grassi (può verificarsi rachitismo). È possibile riscontrare displasia renale con reni piccoli (comune nella sindrome di Alagille di tipo 2) e ipotiroidismo.
Sindrome di Rubinstein-Taybi
La sindrome di Rubinstein-Taybi è una sindrome malformativa rara, caratterizzata da anomalie congenite (microcefalia, facies caratteristica, pollici e alluci larghi e ritardo della crescita postnatale), bassa statura, disabilità cognitiva e disturbi comportamentali.
Sindrome di WAGR
WAGR è l’acronimo per la sindrome tumore di Wilms – aniridia – anomalie genitourinarie – ritardo mentale. Il tumore di Wilms o nefroblastoma è il tumore renale più frequente nei bambini, responsabile del 6-8% dei cancri pediatrici. Circa l’1% di questi tumori si associa a aniridia. La prevalenza della sindrome WAGR è inferiore a 1 su 100.000 nati. La sindrome è associata a un rischio aumentato di tumore di Wilms, che si può evidenziare a qualsiasi età, e a aniridia totale o parziale, con possibile glaucoma o cataratta, disturbi genitourinari, che variano da ambiguità sessuale a ectopia dei testicoli e ritardo mentale variabile. In un bambino sono stati riportati segni oculari atipici quali microftalmia bilaterale, anomalie corneali e agenesia della camera anteriore sinistra con disfunzione retinica.
Sindrome di Potocki-Shaffer
La sindrome di Potocki-Shaffer è caratterizzata da esostosi multiple, forami parietali, fontanella anteriore ampia e, occasionalmente, deficit cognitivo e lievi dismorfismi cranio-facciali. Fino ad oggi sono stati osservati 23 pazienti provenienti da 14 famiglie. La sindrome è causata da una delezione di geni contigui sul braccio corto del cromosoma 11p11.2.
Sindrome di Miller-Dieker
La lissencefalia tipo 1 da anomalie di LIS 1 o sindrome di Miller-Dieker (MDS) è una sindrome da delezione di geni contigui sul cromosoma 17p13.3, caratterizzata da lissencefalia classica (lissencefalia tipo 1) e segni facciali caratteristici. Possono fare parte di questa condizione altre malformazioni congenite. La MDS è, senza dubbio, una malattia rara osservata fino ad oggi in 11, 7 nati per milione di nati vivi, anche se la prevalenza e l’incidenza sono probabilmente sottostimate. I bambini con MDS presentano un grave ritardo di crescita, di solito associato a epilessia e disturbi dell’alimentazione. La lissencefalia rappresenta lo spettro fenotipico estremo di un quadro di agiria generalizzata o agiria e pachigiria frontale. In quasi il 100% dei pazienti sono presenti delezioni visibili o submicroscopiche della regione 17p13.3, che coinvolgono il gene LIS1. La presa in carico dei bambini con MDS è sintomatica.
Sindrome da delezione 1q21.1
La sindrome da microdelezione 1q21.1 è una malattia da delezione ricorrente, descritta di recente, caratterizzata da un quadro clinico variabile, diverso da quello osservato nella sindrome trombocitopenia-aplasia del radio (TAR). È stata descritta in 46 pazienti. Il fenotipo clinico è estremamente variabile; i segni più comuni, ma incostanti, sono la microcefalia, il ritardo dello sviluppo o il lieve deficit cognitivo, i dismorfismi facciali, peraltro poco marcati, e le anomalie oculari. Non sono comuni le malformazioni congenite. Di rado sono stati descritti i disturbi dello spettro autistico, la schizofrenia o il deficit dell’attenzione con iperattività.
Sindrome di Kleefstra
La sindrome di Kleefstra (KS) è una malattia genetica caratterizzata da deficit cognitivo, ipotonia infantile, grave ritardo del linguaggio espressivo e facies caratteristica, associati a una serie di segni clinici aggiuntivi.
Sindrome di Phelan-Mcdermid
La sindrome da monosomia 22q13 (sindrome da delezione 22q13.3 o sindrome di Phelan-McDermid) è una sindrome da microdelezione cromosomica, caratterizzata da ipotonia neonatale, ritardo globale dello sviluppo, crescita accelerata o normale, linguaggio assente o gravemente ritardato e dismorfismi. Data la difficoltà della diagnosi clinica e la scarsa disponibilità di test di laboratorio, la sindrome è sotto diagnosticata e la sua reale incidenza non è ancora nota. La delezione ha una frequenza analoga nei maschi e nelle femmine e sono state osservate forme omogenee e in mosaico. Le caratteristiche cliniche comuni comprendono le ciglia lunghe, le orecchie grandi o dismorfiche, le mani relativamente grandi, le unghie dei piedi displasiche, le sopracciglia folte, la dolicocefalia, le guance piene, il naso globoso e il mento appuntito. Il comportamento è simil-autistico con diminuzione della percezione del dolore e frequente masticazione e movimenti della bocca.
Studio cromosomico completo
Rappresenta l’analisi del cariotipo fetale che rileva anomalie del numero dei cromosomi 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 14, 15, 16, 17, 19, 20, 22. In relazione ad eventuali anomalie riscontrate esistono problematiche di particolare gravità che conducono spesso all’aborto.
La fibrosi cistica materna
La Fibrosi Cistica (FC) è una malattia ereditaria a trasmissione autosomica recessiva, multisistemica, in cui il trasporto difettoso del cloro attraverso le membrane cellulari causa la presenza di secrezioni disidratate e viscose con conseguente addensamento di muco nei bronchi, ispessimento del succo pancreatico ed elevati livelli di cloro nel sudore. Gli organi frequentemente interessati sono il fegato, l’intestino, l’apparato riproduttivo e i polmoni dove il muco particolarmente denso porta a gravi problemi respiratori e a conseguenti infezioni. I bambini malati di Fibrosi Cistica hanno 2 genitori che sono almeno portatori sani (o eterozigoti) di una mutazione del gene e, in particolare, tra due genitori portatori sani esiste una probabilità del 25% che il feto possa essere affetto della malattia. Ad oggi sono riconosciute più di 1500 mutazioni del gene della Fibrosi Cistica (CFTR) molte di esse sono estremamente rare ed altre sono ancora sconosciute. Con FetalDNA Cariotipo Plus viene effettuata l’analisi del gene materno attraverso uno screening chiamato di 1° livello che permette di analizzare le mutazioni più comuni e frequenti riuscendo ad identificare circa l’83% dei portatori. La frequenza stimata, nella popolazione italiana, dei portatori sani (spesso inconsapevoli di esserlo) è di 1 su 25 –30, quella dei nati affetti è di 1 su 2500 – 3000.
Metodo di studio: Estrazione del DNA materno, amplificazione specifica del gene CFTR.
Mutazioni analizzate:
711+1G-T, 621+1G-T, 1717-1G-A, 3849+10kbC-T, 2789+5G-A, G542X, G85E, G551D, R553X, N1303K, R117H, R1162X, L1077P, L1065P, W1282X, R347P, I507del, T338I, F508del, 1677delTA, 2183AA-G, S549R, Q552X, 852del22, R1066H, G1244E, 1259insA, D1152H, 711+5G-A, R1158X, 4382delA, 4016insT, A455E, 1706del17, I502T, 3199del6, S912X.
Note: La frequenza cumulativa, nell’ Italia Centrale, delle mutazioni analizzate mediante questa analisi è di circa l’ 83 % (Genetic History of Cystic Fibrosis Mutation in Italy. I: Regional Distribution: Ann. Human.Genet., 1997).
Risultati ottenibili:
Eterozigote o portatore sano: indica che è stata riscontrata, nel gene CFTR materno, una mutazione tra quelle analizzate. In questi casi verrà consigliata una consulenza genetica con uno dei genetisti del nostro centro per meglio comprendere il dato analitico e per consigliare eventuali approfondimenti diagnostici (ad esempio analisi del DNA paterno).
Negativo: indica che non è stata riscontrata, nel gene CFTR materno, nessuna delle mutazioni tra quelle analizzate. Questo non esclude che la madre sia portatrice sana per mutazioni rare e non presenti tra quelle indagate; tale rischio residuo è approssimativamente di 1/150. È possibile ridurre tale rischio residuo mediante un approfondimento detto di 2° livello che prevede l’analisi dell’intero gene CFTR tramite tecnologia NGS (Next Generation Sequencing) che esclude il 99% delle mutazioni ad oggi note.